



业务咨询

![]() 发布时间:2023-11-03
发布时间:2023-11-03
![]() 环特生物
环特生物
![]() 浏览次数:4625
浏览次数:4625

编者按
由于复杂的发病机制和高度可变的个体药物易感性,使得药物诱导性肝损伤(DILI)的临床前鉴定成为了一项重大挑战。因此急切需要建立一个能够在体外预测DILI的有效模型。
今天,我们回顾了一项于2021年发表在《Gastroenterology》的经典研究——《High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell–Derived Organoids》,该研究利用人多能干细胞诱导的肝类器官进行高保真药物性肝损伤筛查。
一、研究背景
由于在最初的筛选中发现的候选药物失败,制药行业每年在药物开发上损失数十亿美元,近三分之一的药物被撤出市场。此外,尽管有很好的疗效,但候选药物的失败会导致患者治疗机会减少。临床前研究通常将药物的体外评估作为主要的疗效筛选标准,以确定合适的化合物,然后再进行体外和体内安全性研究,以评估其代谢和毒理学机制。这种低效率的方法可以解释为在评估人类药物性肝损伤(DILI)时缺乏生理学相关的临床前模型,因此迫切需要开发一种体外筛选模型来评估大量不断增长的药物化合物文库。
肝原代细胞是一种高度极化的代谢细胞类型,形成具有微绒毛通道的胆管结构,将外周循环与胆汁酸分泌途径分开。DILI的最上游环节包括肝细胞对药物或其反应性代谢物的解毒,并通过转运体,如多药耐药相关蛋白(MRP)转运体)排泄到胆管,这表明需要重建这些独特的组织结构,作为体内肝细胞预测DILI病理的关键特性。
然而,目前使用分离的原代人肝细胞或肝细胞系的简化培养模型与体内生理学之间的药物毒性特征存在相当大的差异,导致药物转化失败或停滞,例如,曲格列酮、奈法唑酮和托尔卡酮。因此,毒理学特性的测定主要依靠动物作为药物开发的重要步骤;然而,由于人类和动物在生理上的显著差异,对人类结果的保真度明显不足。
本文的研究人员最近报道了一种从多能干细胞(PSCs)生成人类肝脏类器官(HLOs)的方法,该方法具有模拟炎症疾病的潜力。然而,功能性胆小管样结构,作为模拟胆汁排泄缺陷的重要组成部分,是否形成并可用于药物毒理学分析仍是一个谜。此外,在最大限度地减少批次差异的同时,在转化为临床前研究之前,提高分析通量将是至关重要的。
在此,本文的研究人员使用稳定可扩增的诱导PSC(iPSC)和胚胎干细胞。本文的研究人员建立了一种基于实时成像的动态检测方法,可以通过敲除胆汁酸转运体基因来阻止改良HLOs中胆汁酸的摄取和排泄。该检测平台适用于大规模的化合物筛选和解释238种具有多重读数的药物。此外,HLOs被证明可以模拟基因型特异性对波生坦诱导的多个iPSC的胆汁淤积的易感性。这种名为肝类器官毒性筛查(LoT)的强大检测方法提供了HLOs中开发的功能读数,将促进诊断、功能研究、药物开发和个性化医疗。
二、主要研究成果
1、人诱导多能干细胞诱导肝类器官的生成与表征
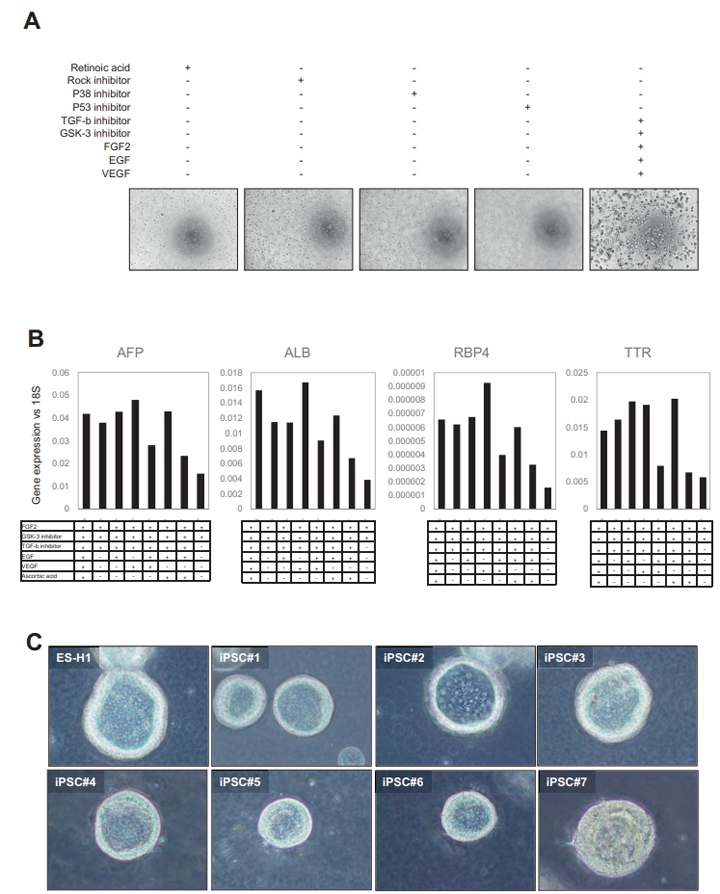
本文的研究人员首先建立了一种利用hiPSC衍生的前肠球体获得大量均匀类器官的新方法(图1A)。简单地说,前肠细胞在第7天消化成单个细胞。在这个阶段,游离的前肠细胞可以冷冻保存在-80℃。将新鲜或解冻的细胞包埋于Matrigel中,然后在由FGF2表皮生长因子、血管内皮生长因子、GSK抑制剂(CHIR99021)和转化生长因子-B抑制剂(A83-01),与抗坏血酸(补充图2A和B)组成的类器官形成培养基中培养4天。第二天,本文的研究人员将培养物暴露在维甲酸(RA)中4天,维甲酸促进了胆管和胆管周鞘的形成。
接下来,本文的研究人员切换到肝细胞成熟培养基。高效地生成具有腔内结构的类器官(图1B),在5个因子的培养基中诱导的HLOs数量远大于常规因子的培养基中诱导的,仅用RA处理(图1C和D)。本文的研究人员证实HLOs是在5个因子下由多个iPSC细胞系形成的((补充图2C)。此外,本文的研究人员证明HLOs在含有5个因子的类器官形成培养基中培养,然后在RA培养96小时,具有最高的白蛋白分泌效率(图1E)。
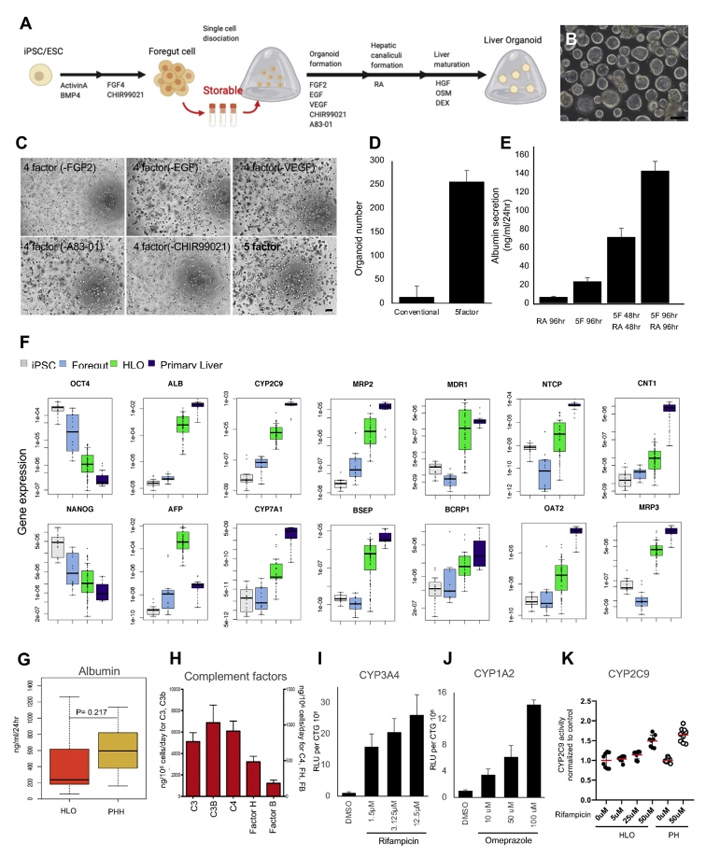
图1 从可冻存的psc来源的前肠细胞生成HLOs
2、人肝类器官的基因表达和功能分析
采用定量聚合酶链反应(qPCR)分析了HLOs中与肝功能倾向相关的代表性基因的表达,以揭示其肝脏谱系。肝脏标志物基因如白蛋白(ALB)、甲胎蛋白(AFP)、细胞色素P(CYP)450家族2亚家族C成员9 (CYP2C9)和CYP7A1在HLO中表达上调(图1F)。多药耐药相关蛋白2(MRP2)、胆汁盐输出泵(BSEP)、多药耐药1(MDR1)、BCRP1、纳米-牛磺酸胆酸共转运多肽(NTCP)、有机阴离子转运蛋白2 (OAT2)、浓缩核苷转运蛋白1(CNT1)、MRP3等极性和转运蛋白活性相关基因的表达也有所增加。此外,这还伴随着与未分化状态相关的基因(如NANOG和OCT4)的逐渐减少。NANOG、OCT4、ALB、CYP2C9、MRP3和TDO2在6种不同供体的HLOs中的基因表达具有可比性((补充图3A)。
HLOs的白蛋白分泌能力如图1G所示。HLO和原代肝细胞白蛋白分泌水平分别为488.2±517.5 ng/mL/d和630.49±292.5 ng/mL/d,差异无统计学意义。虽然HLOs和原代肝细胞的白蛋白分泌水平相当,但两者的白蛋白基因表达不同,这可能是由于HLOs和原代肝细胞的细胞组成不同所致。
酶联免疫吸附法也证实了HLO培养上清中存在补体因子等肝细胞特异性蛋白(图1H)。
最后,在功能水平上,本文的研究人员展示了HLO分别经利福平和奥美拉唑处理后的主要CYP诱导反应(图1I和图J)。此外,本文的研究人员还检测了HLO中CYP2C9的诱导性,经利福平处理后,HLO中CYP2C9的反应性与原代肝细胞相当(图1K)。
3、人肝类器官单细胞RNA测序分析
为了与原代肝细胞的分化状态进行比较,本文的研究人员利用单细胞RNA测序(scRNA-seq)技术对第20天HLOs中的HLO来源细胞(5177细胞)进行了测序,并与5个独立供体来源的原代人肝来源细胞[7]进行了整合分析(图2A和附图4)。t -分布随机近邻嵌入分析表明,HLOs中存在由实质细胞(74.41 %)和非实质细胞(25.59 %)组成的不同种群(图2B和图C),在原代肝细胞中表达肝星状细胞、门静脉内皮细胞和胆管细胞的特征性标志物,如本文的研究人员之前的方法所见。
接下来,本文的研究人员研究了与近中心的、汇管区、间充质和内皮相关的基因表达,并与人原代肝细胞(补充图4A)进行了比较。其中,虽然HLO来源的细胞包括非实质细胞群,但肝细胞的亚群(称为肝细胞样1、2细胞)与原代肝细胞几乎相同,其余半数细胞代表肝母细胞样状态。在间充质和内皮的基因集合中,HLOs中的基因表达与人原代肝细胞具有相似性。
为了进一步探讨肝细胞的特性,本文的研究人员从HLO和人原代肝细胞样本中分离出肝细胞群体,并进行t分布随机邻嵌入分析。本文的研究人员发现88.99%的肝细胞样2细胞位于门静脉周围原代肝细胞群中,而27.91%的肝细胞样1细胞位于中心周围原代肝细胞群中(图2D和E)。
3区(肝脏的小叶中心区域)在药物代谢和解毒方面发挥作用。最近在人类scRNA-seq[7]中报道的4个带状近中心的标记物的表达水平与原代肝细胞相似,其中包括一个关键的药物代谢酶CYP2C9(图1F)。通路分析表明,在中枢周围和门户周围的高富集的基因组分别与HLOs患者的脂质/药物代谢、CYP450和胆固醇生物合成过程相关。在HLOs的这些通路基因表达水平与一些原发性肝脏相似(补充图4B)。总的来说,本文的研究人员的HLO模型包含了不同的和区域性的肝细胞群,在一定程度上模拟了原发性成人原代肝细胞的特征。
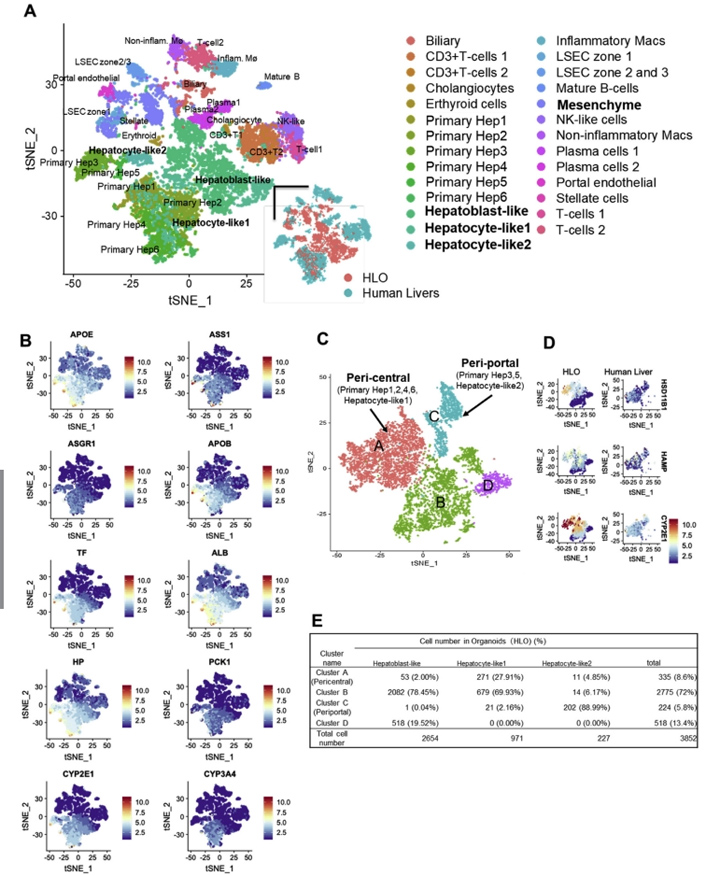
图2 HLOs与人原代肝细胞的scRNA-seq分析
4、人类肝脏类器官的微观解剖特征
免疫组化分析显示HLO上皮细胞白蛋白和IV型胶原染色(图3A)。此外,ZO-1、MRP2、BSEP、F-actin和MDR3的免疫染色表明,这些蛋白在染色肝细胞核因子4a(HNF4a)染色阳性的情况下优先定位于腔内区域。CYP7A1在HLO中也呈阳性染色。
胆小管是肝内最小的分泌通道,胆小管管腔由相邻肝细胞相对质膜的改良的顶端区域形成的空间组成。此外,它被紧密的连接复合体所划分,微绒毛位于小管腔的内部。同样,HLO的透射电镜分析证实肝细胞样细胞之间存在胆管样结构(图3B,左)。
透射电镜分析还显示,HLO含有指向管腔的微绒毛(图3B,右)。免疫组织化学分析显示,MDR3、MRP2、BSEP和ZO-1染色了腔内内膜,表明这些HLOs具有极化特征。与这些解剖特征一致,qPCR分析显示HLOs具有BSEP和NTCP基因表达(图1F)。因此,HLOs包含从内腔中分离出来的极化的人肝细胞,被小管样结构包围,这反映了一种独特的微观解剖结构,类似于活体肝组织。
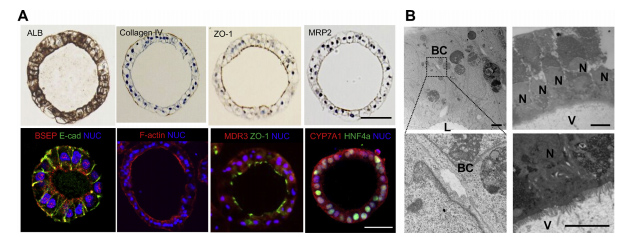
图3 HLO的结构剖面
5、人肝类器官胆汁酸的产生和转运特性
接下来,为了确定胆汁酸的生产能力,本文的研究人员对从类器官培养中收集的腔内液进行了胆汁酸酶联免疫吸附试验。如图1F和3A所示,催化胆固醇分解代谢和胆汁酸合成第一步的CYP7A1在HLOs中分别表达阳性,染色阳性,提示HLOs中胆汁酸合成途径被激活。腔内液总胆汁酸池的水平为26.7μg/d/106个细胞(单个直径为200 μm的HLO为125 μmol/L)(图4A),令人惊讶的是,胆汁酸浓度与之前报道的夹层培养的原代肝细胞相当(40μg/d/106个细胞,培养上清为10 μmol/L)。胆汁酸排泄是胆汁流动的主要决定因素,因此,该系统的缺陷可能导致胆汁分泌受损(胆汁淤积),并与各种肝脏疾病病理相关。
位于肝细胞顶端(小管)膜的外排转运蛋白在肝脏清除许多内源性和外源性化合物(包括药物和代谢物)中发挥重要作用。BSEP和MRP2调节人体小管胆盐运输。由于图3A中胆汁转运关键蛋白的阳性表达,本文的研究人员接下来想知道HLO是否能主动将胆汁酸转运到其腔内。为了监测HLOs中胆汁运输活性的动态变化,采用双醋酸荧光素(FD),在体外研究了从窦膜到胆小管膜的主动运输机制[11]。用FD孵育的HLOs显示,FD在45分钟内被输送到HLOs内部,并依次改变强度(图4B)。此外,发现荧光胆汁酸CLF和胆盐类似物胆酰酰氨基荧光素(分别是胆汁酸运输活性指标和胆盐类似物可重复地从体外排泄并积聚到HLOs腔内(补充图5A和B)。
为了更深入地确定特异性,本文的研究人员利用CRISPR-Cas9基因编辑开发了一个iPSC系,该系携带一个去功能化的BSEP等位基因(图4C)。虽然BSEP突变的iPSC-HLO的形态与对照HLO相似(图4D),但与亲代对照HLO相比,BSEP突变的HLO未能积累荧光胆汁酸(图4E)。类似地,化学BSEP抑制剂sitaxentan在可行剂量下抑制正常HLOs中的CLF运输(图4F和G)。这些数据表明,HLOs具有从外部摄取胆汁酸并将其排出HLO内部的能力。因此,HLOs不仅具有小管样形态,而且具有胆汁酸产生和分泌活性。
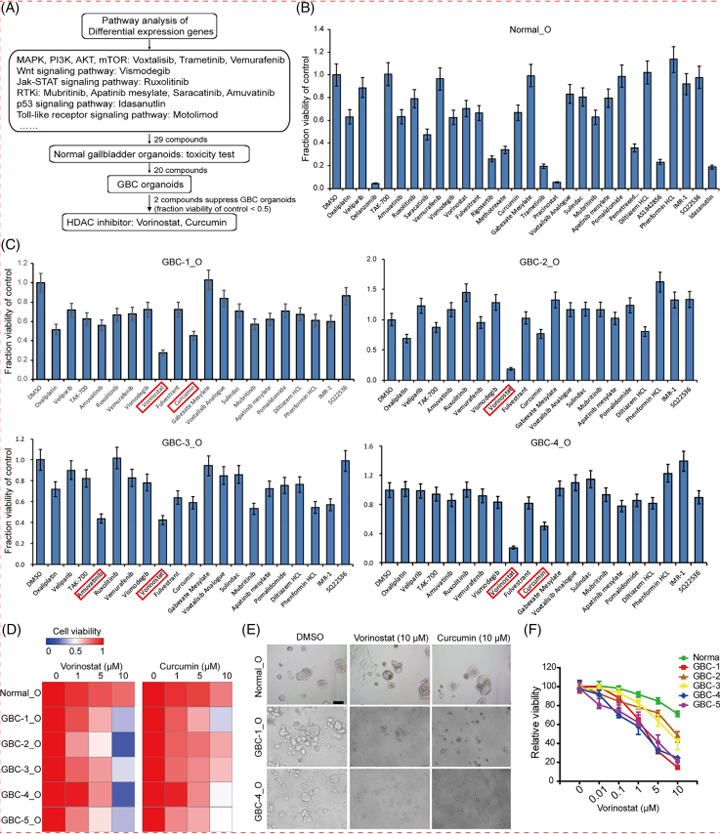
图4 胆汁在HLO中的转运特性
6、人肝类器官高通量药物诱导胆汁淤积评价
在药物发现过程中,在DILI研究的早期阶段,功能和可量化的分析对先导化合物的生成至关重要。然而,目前的模型的读数过于简单,例如,尽管涉及多种因素,如胆汁运输缺陷,但只有生存能力。为了解决这些限制,本文的研究人员开发了基于384孔的高速实时成像平台,本文的研究人员称之为基于HLO的高通量毒性筛选(LoT)模型(图5A)。本文的研究人员的LoT系统可以通过独特的成像算法(图5B)对每孔15-20个类器官进行评估,并对238种上市药物进行了功能验证,其中包括32种阴性对照药物和206种报告的DILI化合物,它们具有4种不同的浓度,基于双重数据:生存能力和胆固醇抑制功能。
在系统中,观察到代表性的抑胆药对CLF转运的抑制作用。首先,与之前的报道相比,本文的研究人员筛选了对多剂量治疗反应的生存能力(补充表1)。为了评估LoT系统的可预测性,本文的研究人员提取了补充图1中列出的每种化合物的临床最大药物浓度(Cmax)值,并在最接近Cmax的浓度下分析数据。结果:敏感性和特异性分别为88.7%和88.9%(或69.0%和100%);并提供了与先前基于原代肝细胞模型的报告相当或更高的值(补充图2和图5C)。
有趣的是,吲哚美辛(Cmax: 8.3 μmol/L)和齐流通 (Cmax: 13.12 μmol/L)分别在10 μmol/L和100 μmol/L的细胞毒实验中成功检测到毒性,而这两种药物在其他平台上很难检测到[19,20]。其中,已知诱导胆汁淤积的药物倾向于降低CLF强度,这表明可以在类器官成像分析中模拟显著的胆汁转运抑制作用(补充表3和图5D)。总之,本文的研究人员在这里开发的LoT系统由于其吞吐量和与人类数据的相关性,有可能用于早期药物发现过程。
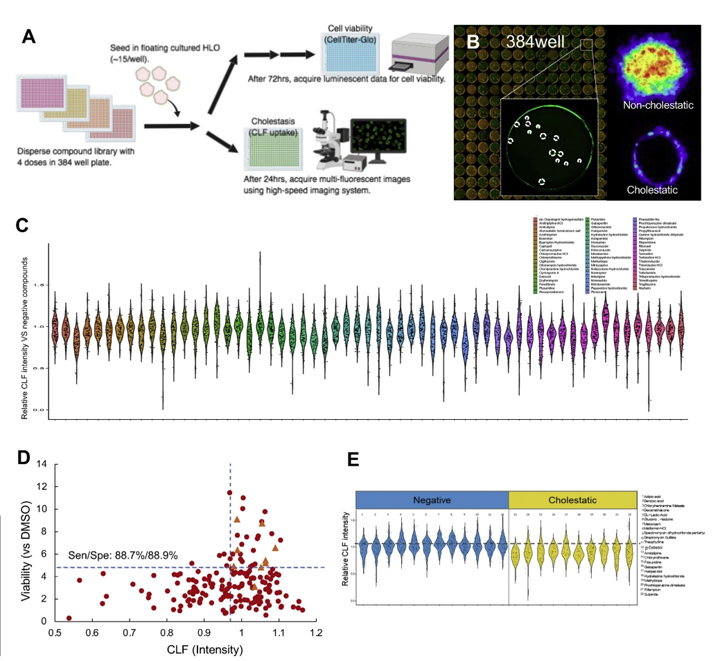
图5 LoT筛选可识别具有潜在毒性的化合物
7、基于肝类器官的高通量毒性筛选系统对药物性肝损伤化合物机制分类的研究
接下来,本文的研究人员着手建立一个使用类器官的深入调查模型。为了监测线粒体膜电位(MMP)线粒体健康以及胆汁运输,本文的研究人员在完整的类器官中重复读数。在确定了10种美国食品和药物管理局批准的不影响生存能力的药物的最佳剂量后(补充结果和补充图6),本文的研究人员用MMP量化线粒体健康评估。用训练化合物(TCs)治疗24小时后,观察到托卡酮(2-8倍变化,P < 0.01)、双氯芬酸(7- 13倍变化,P < 0.05或0.01)、环孢素A(3- 7倍变化,P < 0.01)和奈法唑酮(4- 42倍变化,P < 0.01)治疗后的MMP的剂量依赖性增加(图6A-C)。
此外,曲格列酮也增加HLO患者的MMP(3- 5倍变化,P < 0.05),但未观察到剂量依赖性。另一方面,在波生坦、恩他卡彭和吡格列酮治疗后,即使多剂量也没有明显观察到MMP的增加。
人类DILI的严重表现是多因素的,并且与已知的DILI机制(如线粒体和BSEP抑制)的组合高度相关。本文的研究人员进一步分析了存活、胆汁淤积和线粒体应激之间的关系。为了将本文的研究人员的毒性试验与传统的体外试验系统进行比较,本文的研究人员根据已发表的关于原代肝细胞系统的胆汁淤滞毒性和线粒体应激的报告,选择了TCs及其浓度。值得注意的是,与托尔卡彭、双氯芬酸和波生坦相比,24小时双效药物(胆碱抑制和线粒体应激),如环孢素A、曲格列酮和奈法唑酮,在72小时显著降低细胞活力(图6D和补充图6A)。
这些数据与临床数据相当,表明双重效力与DILI的严重程度高度相关,这与以前的报告一致。此外,本文的研究人员还注意到,130μmol/L的恩他卡彭处理降低了类器官活力(从24小时的85%降至72小时的64%)。恩塔卡彭需要广泛的结合血浆蛋白,主要是白蛋白,诱导DILI。利用LoT系统进一步研究恩他卡酮除胆汁淤积和线粒体健康外的毒性机制将是有趣的,因为毒性机制尚未明确。综上所述,LoT系统是DILI主要机制分类的有用人体模型系统,也是进一步描述未知复杂机制的测试平台。
众所周知,DILI的发病率常常与许多宿主因素相混淆。例如,越来越多的证据表明,当与某些药物(如对乙酰氨基酚)联合使用时,肥胖和非酒精性脂肪性肝病会大大增加啮齿动物和人类的肝毒性风险。因此,即使患者处于亚临床阶段,在如此“脆弱”的情况下,预测DILI的潜力似乎很重要。
在这里,本文的研究人员建立了脂毒性类器官模型通过暴露于不饱和脂肪酸油酸中(图6E)。在油酸处理3天后,HLOs中的脂质积累非常强烈(图6E)。脂肪酸的氧化是活性氧(ROS)的重要来源,它导致5′-三磷酸腺苷和烟酰胺二核苷酸的耗竭,并诱导脂肪肝DNA损伤。与此一致的是,脂质处理的HLOs中ROS的生成增加(图6F和补充图7A)。此外,脂肪酸诱导肝脏线粒体大量肿胀(图6F和补充图7B),类似于已发表的表型。
由于这种脂毒性类器官模型是ROS增加的脆弱状态,因此采用2噻唑烷二酮、曲格列酮(0-50μmol/L)和吡格列酮(200μmol/L)处理HLOs 24小时,并评估细胞活力。本文的研究人员观察到,在脂毒性条件下,使用曲格列酮治疗后,由于细胞死亡导致类器官大量碎裂(补充图8),随后的细胞活力分析证实了这一点(与对照组相比,细胞存活率为40%,P < 0.05)(图6G)。由于单独使用吡格列酮治疗不会影响有或没有脂毒性的HLO的生存能力,因此脂毒性HLO可能突出DILI易损性,具有阳性和阴性的预测能力(图6G)。
接下来,本文的研究人员研究了HLOs是否可以使用具有治疗潜力的化合物从DILI样疾病中恢复。为了证明这一概念,本文的研究人员使用抗氧化剂N-乙酰半胱氨酸(NAC)来抑制ROS的产生,这是基于文献报道,静脉注射NAC可以提高非对乙酰氨基酚相关急性肝衰竭患者的生存率,并降低曲格列酮诱导的细胞毒性[28]。正如预期的那样,NAC显著提高了细胞活力,这表明即使在脆弱条件下,NAC也可以挽救HLOs中的细胞死亡(图6G和补充图8)。该LoT系统可能作为临床前工具,在多药方案下识别缓解DILI样疾病的有效化合物。
8.CYP2C9*2诱导的多能干细胞肝类器官特异性波生坦诱导的胆汁淤积
由于药物诱导的事件是高度可变的,基于ipsc的类器官方法有望用于个体敏感性的潜在评估[4]-。为了确定本文的研究人员的系统的临床相关性,本文的研究人员使用了影响药物诱导的胆汁转运抑制潜力的药物基因组学见解。
本文的研究人员对8种不同的iPSC系进行了基因分型,其中有或没有众所周知的易感基因变异(即CYP2C9*2活性中间体),以波生坦诱导DILI,并比较了HLO系的胆汁抑制潜能。有趣的是,CLF向HLOs的排泄严重受损(阳性率:CC,阳性17.1%;在CY2C9*2携带者的HLOs中,波生坦的阳性率为70.8%,而在3种不含CYP2C9*2的ipsc衍生的HLOs中则没有(图7A-E)。
这些结果表明,基于类器官的胆汁淤积试验对CYP2C9介导的药物性胆汁淤积变异有阴性和阳性的预测,正如在人类中看到的那样。这些结果表明,以类器官为基础的胆汁淤积试验预测了药物引起的胆汁淤积的人类遗传变异的某些方面。
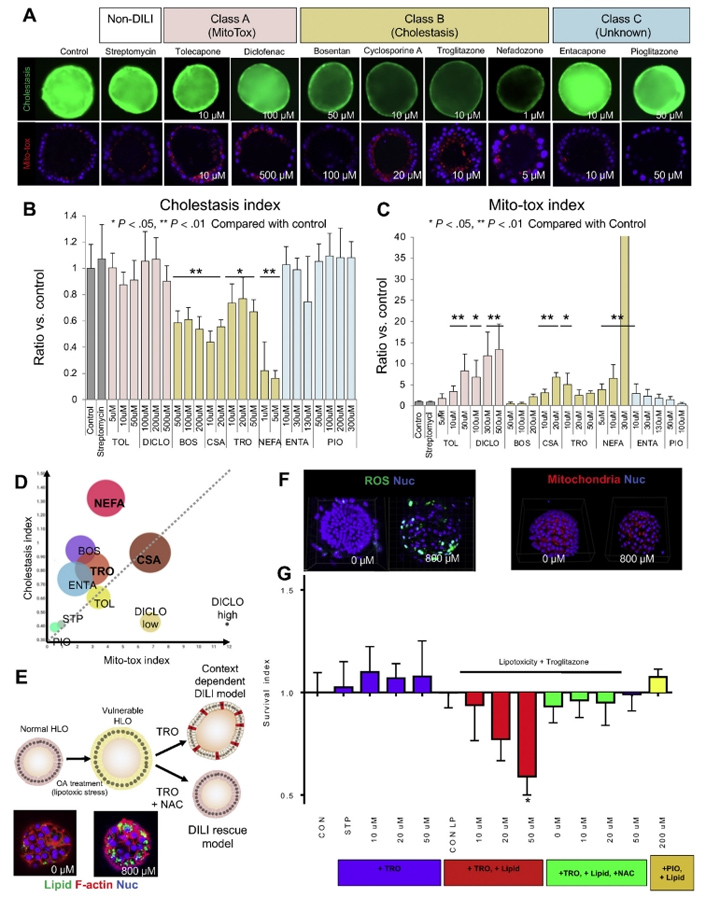
图6 采用DILI HLO模型的机械毒理学方法
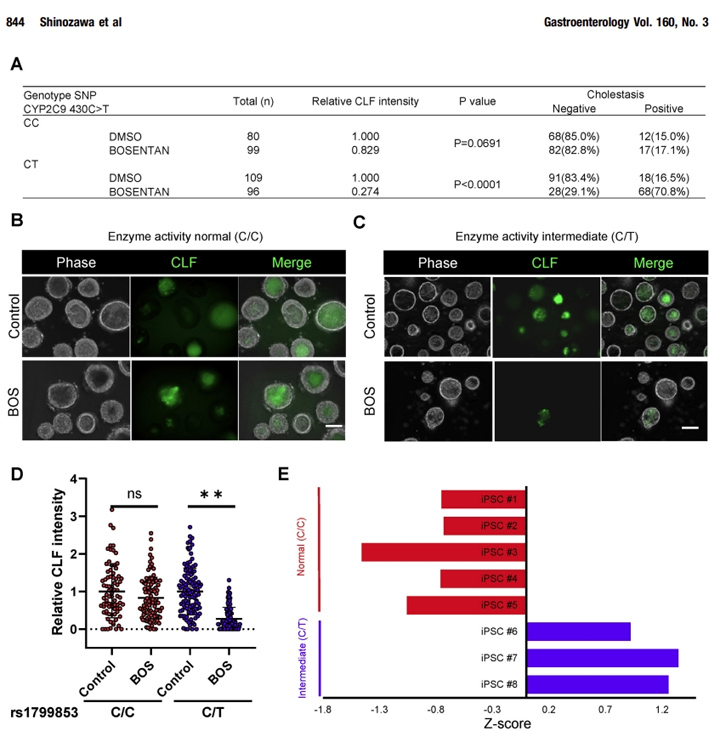
图7 波生坦诱导的胆汁淤积是CYP2C9*2 HLO特异性的
三、补充结果
9.大型类器官毒性系统对药物性肝损伤化合物机制分类的回顾
由于线粒体毒性在与DILI发病相关的多种机制中在DILI中起核心作用,因此用MMP指数来描述HLO患者的线粒体健康状况是有意义的[30]。为了监测MMP线粒体健康以及胆汁运输,本文的研究人员对完整类器官的读数进行了多重处理。在测试化合物阵列之前,本文的研究人员将MMP应用于具有FD的类器官中以检测胆汁运输活性,而不是荧光标记的胆汁酸CLF和胆酰甘酰胺荧光素,因为FD可以平行获取MMP。与FD孵育的HLO在与MMP激活一致的较短动力学(20-45分钟)中表现出相同的表型(图4B)。然后,本文的研究人员做了注释美国食品和药物管理局批准了10种基于fd的检测和MMP检测的药物。
首先,虽然本文的研究人员找到了9种化合物的最佳预死剂量,但在本文的研究人员的测试范围内,胺碘酮对HLOs具有显著毒性(补充图6A);因此,本文的研究人员在进一步的潜在DILI评估研究中排除了胺碘酮,以避免细胞死亡引起的继发性改变引起的噪音。根据DILI机制将9种tc分为3类:无胆汁淤积的DILI化合物(A类)、有胆汁淤积的DILI化合物(B类)和未报道为DILI化合物的化合物(C类)(Supplementary Figure 6B)。
为了量化FD排泄的抑制电位,本文的研究人员通过ImageJ量化了每个类器官内外的荧光强度比(补充图5C)。为了确定最佳测量时间,本文的研究人员使用CSA测试了FD转运抑制。在FD治疗后5分钟,CSA治疗24小时组与对照组(DMSO)相比显著下降(与对照组相比下降0.4)(补充图5C)。用同样的方法,本文的研究人员评估了9种不同浓度的TCs, B类化合物的FD外排明显减少(P < 0.01或0.05);波生坦、CSA、曲格列酮和奈法唑酮与临床观察结果相似,而在A类和C类化合物中没有观察到这种抑制作用(图6A和B)。

补充图1 用于定量的HLOs图像。图像显示HLO大小和形状均匀
补充图2 从多个多能干细胞生成HLO的稳健方案的发展
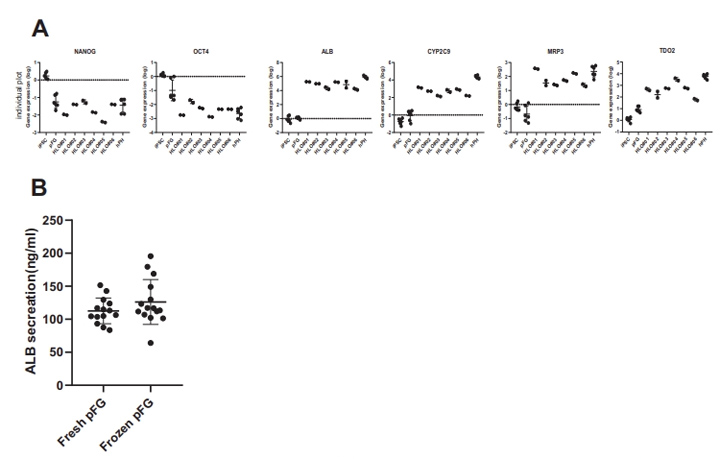
补充图3 来自多个psc的HLO分析
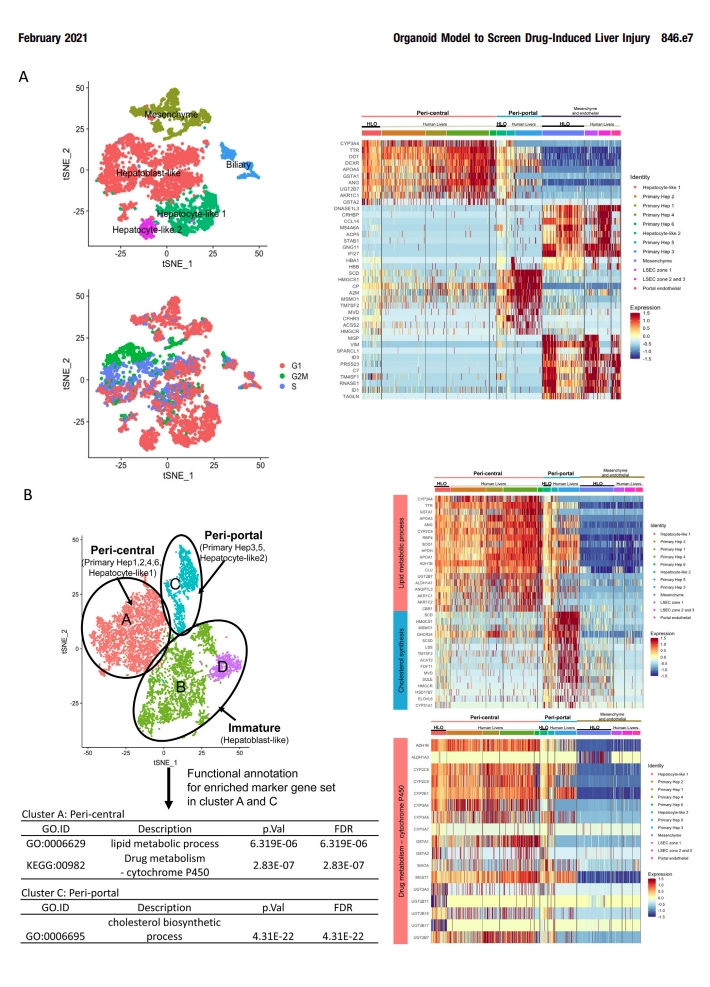
补充图4 HLOs的scRNA-seq分析
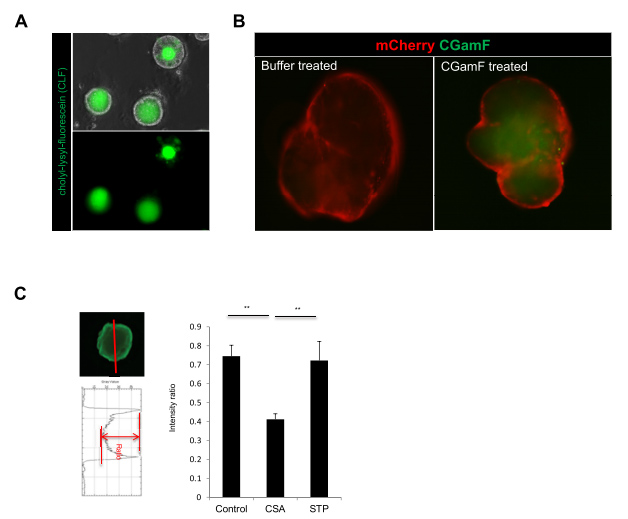
补充图5 用探头观察胆汁转运功能
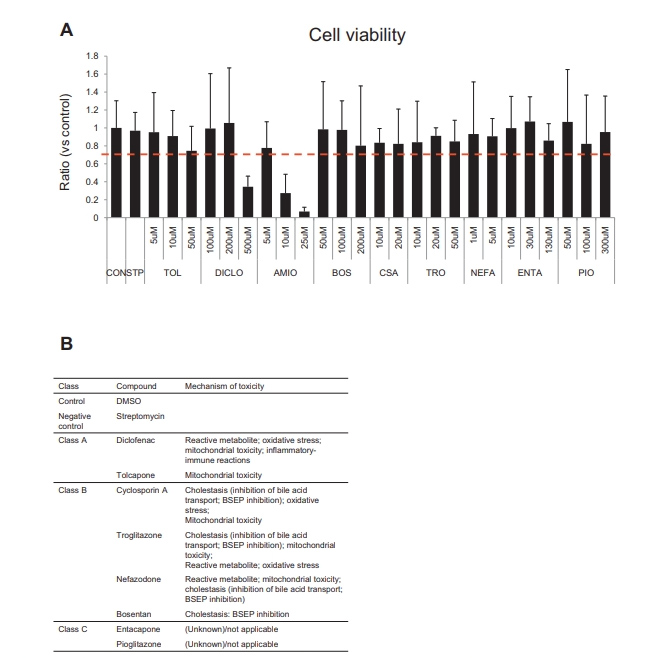
补充图6 10种化合物处理后24小时的细胞活力

补充图7 脂毒性肝类器官中ROS的产生和线粒体形态的改变

补充图8 脂毒性类器官模型。无脂质堆积模型的形态学变化(上表)以及脂质和药物治疗情况
四、编者点评
人类成体干细胞衍生的肝类器官可以恢复成熟的肝细胞特征,从而可以在体内损伤的肝脏中重新填充。细胞内具有管腔和胆管样结构的HLOs的形态与原代类器官既有异同又有相似之处。例如,iPSC-HLO显示具有大管腔的中空样结构,而原代组织包含具有小管鸡丝状外观的细胞索。
然而,这两个系统都包含管状管腔,管状管腔是由相邻肝细胞相对质膜的修饰的顶端区域形成的空间,以及位于管腔内部的紧密连接复合物和微绒毛的分布。基因编辑和药物抑制剂试验证实胆汁酸类似物内化到HLO中使用BSEP,这是一种已知的胆汁酸排泄转运蛋白。考虑到通过scRNA-seq, HLO中一半的肝细胞样细胞显示不成熟状态,与成人肝组织来源的基于类器官的方法相比,psc来源的HLO表现出相对不成熟的转录组特征,类似于其他已发表的研究。有趣的是,即使在妊娠早期,人类胎儿肝脏也已知具有功能性药物代谢酶系统,这是人类而非动物特有的独特特征。
此外,本文的研究人员的scRNA-seq鉴定的HLO群体亚群代表1区(肝脏门静脉周围区)和3区(肝脏小叶中心区)肝细胞样细胞。HLO中未成熟和成熟的带状肝细胞之间的差异特征保证了未来的研究,以确定信号传导逻辑,以继续HLO中肝细胞样细胞的成熟。
LoT测定的主要优点包括使用患者的iPSC,前肠阶段的可储存特性,测定通量,以及用于分析其他因素(如线粒体应激)之间相互作用的多路读数。如上所述,回顾性研究表明,多种细胞应激电位与DILI[21]的发病率相关,LoT试验显示的结果与该研究相当,因为细胞活力降低依赖于双重读数:线粒体和胆汁沉积应激。
氧化应激在细胞死亡中起重要作用,并与胆汁淤积性肝损伤的发展有关。在胆汁淤积期间,疏水胆汁酸在细胞内积聚,干扰正常的线粒体电子传递,抑制呼吸复合物I和III的活性,从而减少腺苷5′三磷酸的合成,导致线粒体功能障碍诱导的细胞凋亡。
与这些发现一致,本文的研究人员对这两个读数的相关分析表明,与线粒体应激相比,胆汁淤积应激是肝损伤的更主要因素。值得注意的是,与基因变异相关的对波生坦的不同易感性被本文的研究人员使用多供体iPSC衍生的HLOs的LoT系统重演。由于原代肝组织取样困难且效率低,特别是来自无疾病的健康个体的34例,因此,初步证明健康供者来源的iPSC来源的HLOs将是精准研究的有价值的工具CYP2C9*2载体鉴定。更重要的是,最近发展起来的具有基因测序数据的大规模iPSC库将使本文的研究人员能够建立一个与基因组分层相结合的可想象的群体。LoT可以作为制药业的一项令人兴奋的战略,为最大限度地减少DILI的可能性提供必要的见解。
LoT系统目前是一个过度简化的模型,缺乏适应性免疫成分。因此,为了充分预测其他类型的DILI,包括在患者中观察到的特异性DILI和免疫介导性DILI,额外的修改是至关重要的。此外,本研究是在单一机构进行的。为了便于应用于临床前毒理学分析,基于384孔的LoT筛选试验需要在多个独立机构进行盲法化合物试验验证,以进一步提高可重复性和可信度。尽管LoT检测显示在>8名供者中CYP2C9基因变异依赖性胆汁淤积加剧,但这一解释不能排除供者依赖性差异的可能性,而不是基因型依赖性。为了证明两者之间的因果关系,在iPSC中使用等基因碱基编辑方法将是非常有用的。
作为健康美丽产业CRO服务开拓者与引领者、斑马鱼生物技术的全球领导者,环特生物搭建了“斑马鱼、类器官、哺乳动物、人体”四位一体的综合技术服务体系,开展科研服务CRO、智慧实验室建设和精准医疗三大业务。目前,环特类器官平台已开发多种生理类器官模型,如肝、肺、肾、IPSC神经、血脑屏障模型等,欢迎有需要的读者垂询!
参考文献
1.Takebe T, Taniguchi H. Human iPSC-derived miniature organs: a tool for drug studies. Clin Pharmacol Ther 2014;96:310–313.
2.Yang K, Woodhead JL, Watkins PB, et al. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin Pharmacol Ther 2014;96:589–598.
3.Leslie EM, Watkins PB, Kim RB, et al. Differential inhibition of rat and human Naþ-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1)by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 2007;321:1170–1178.
4.Ouchi R, Togo S, Kimura M, et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab 2019;30:374–384.e6.
5.Zhang RR, Koido M, Tadokoro T, et al. Human iPSCderived posterior gut progenitors are expandable and capable of forming gut and liver organoids. Stem Cell Reports 2018;10:780–793.
6.Falasca L, Favale A, Serafino A, et al. The effect of retinoic acid on the re-establishment of differentiated hepatocyte phenotype in primary culture. Cell Tissue Res 1998;293:337–347.
7.MacParland SA, Liu JC, Ma XZ, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383.
8.Tsukada N, Ackerley CA, Phillips MJ. The structure and organization of the bile canalicular cytoskeleton with special reference to actin and actin-binding proteins. Hepatology 1995;21:1106–1113.
9.Ni X, Gao Y, Wu Z, et al. Functional human induced hepatocytes (hiHeps) with bile acid synthesis and transport capacities: a novel in vitro cholestatic model. Sci Rep 2016;6:38694.
10.Kock K, Brouwer KL. A perspective on efflux transport proteins in the liver. Clin Pharmacol Ther 2012;92:599– 612.
11.Yumoto AU, Watanabe S, Hirose M, et al. Structural and functional features of bile canaliculi in adult rat hepatocyte spheroids. Liver 1996;16:61–66.
12.Keitel V, Burdelski M, Vojnisek Z, et al. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology 2009; 50:510–517.
13.Mork LM, Isaksson B, Boran N, et al. Comparison of culture media for bile acid transport studies in primary human hepatocytes. J Clin Exp Hepatol 2012;2:315–322.
14.Khetani SR, Kanchagar C, Ukairo O, et al. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci 2013;132:107–117.
15.O’Brien PJ, Irwin W, Diaz D, et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol 2006; 80:580–604.
16.Vorrink SU, Zhou Y, Ingelman-Sundberg M, et al. Prediction of drug-induced hepatotoxicity using longterm stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol Sci 2018; 163:655–665.
17.Proctor WR, Foster AJ, Vogt J, et al. Utility of spherical human liver microtissues for prediction of clinical druginduced liver injury. Arch Toxicol 2017;91:2849–2863.
18.Xu JJ, Henstock PV, Dunn MC, et al. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol Sci 2008;105:97–105.
19.Fromenty B. Drug-induced liver injury in obesity. J Hepatol 2013;58:824–826.
20.Michaut A, Le Guillou D, Moreau C, et al. A cellular model to study drug-induced liver injury in nonalcoholic fatty liver disease: application to acetaminophen. Toxicol Appl Pharmacol 2016;292:40–55.
21.Aleo MD, Luo Y, Swiss R, et al. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014;60:1015–1022.
22.Oorts M, Baze A, Bachellier P, et al. Drug-induced cholestasis risk assessment in sandwich-cultured human hepatocytes. Toxicol In Vitro 2016;34:179–186.
23.Bort R, Ponsoda X, Jover R, et al. Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther 1999;288:65–72.
24.Fisher A, Croft-Baker J, Davis M, et al. Entacaponeinduced hepatotoxicity and hepatic dysfunction. Mov Disord 2002;17:1362–1365; discussion 1397–1400.
25.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147– 152.
26.Zborowski J, Wojtczak L. Induction of swelling of liver mitochondria by fatty acids of various chain length. Biochim Biophys Acta 1963;70:596–598.
27.Lee WM, Hynan LS, Rossaro L, et al. Intravenous Nacetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009;137:856–864, 864e1.
28.Rachek LI, Yuzefovych LV, Ledoux SP, et al. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol Appl Pharmacol 2009; 240:348–354.
29.Markova SM, De Marco T, Bendjilali N, et al. Association of CYP2C9*2 with bosentan-induced liver injury. Clin Pharmacol Ther 2013;94:678–686.
30.Li N, Oquendo E, Capaldi RA, et al. A systematic assessment of mitochondrial function identified novel signatures for drug-induced mitochondrial disruption in cells. Toxicol Sci 2014;142:261–273.
31.Oorts M, Baze A, Bachellier P, et al. Drug-induced cholestasis risk assessment in sandwich-cultured human hepatocytes. Toxicol In Vitro 2016;34:179–186.
32.Huch M, Gehart H, van Boxtel R, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015;160:299–312.
33.Hu H, Gehart H, Artegiani B, et al. Long-term expansion of functional mouse and human hepatocytes as 3D organoids. Cell 2018;175:1591–1606.e19.
34.Zachos NC, Kovbasnjuk O, Foulke-Abel J, et al. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem 2016;291:3759–3766.
35.Dedhia PH, Bertaux-Skeirik N, Zavros Y, et al. Organoid models of human gastrointestinal development and disease. Gastroenterology 2016;150:1098–1112.
36.Rane A, Sjoqvist F. Drug metabolism in the human fetus and newborn infant. Pediatr Clin North Am 1972;19:37–49.
37.Serviddio G, Pereda J, Pallardo FV, et al. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology 2004;39:711–720.
38.Krahenbuhl S, Talos C, Fischer S, et al. Toxicity of bile acids on the electron transport chain of isolated rat liver mitochondria. Hepatology 1994;19:471–479.
39.Bernardi P. The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim Biophys Acta 1996; 1275:5–9.
40.Koido M, Kawakami E, Fukumura J, et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nature Medicine 2020;26:1541–1548.
