



业务咨询

![]() 发布时间:2023-03-31
发布时间:2023-03-31
![]() 环特生物
环特生物
![]() 浏览次数:7028
浏览次数:7028

原文献:Guan, Y., Enejder, A., Wang, M. et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nat Commun 12, 6138 (2021).
肝纤维化是慢性肝损伤中细胞外基质(ECM)积累所引起的病理变化[1, 2],其导致肝实质细胞丧失,肝功能下降,并有严重的并发症。虽然它最常见的是由病毒感染或慢性酒精暴露引起的后天疾病,但一些遗传疾病也与之密切相关。据报道,活化的肌成纤维细胞及其产生的胶原蛋白是肝纤维形成的必要介质[3-9]。如果不能从根本上改变过量ECM的生成,则没有可用的治疗方法来预防或逆转其进展。
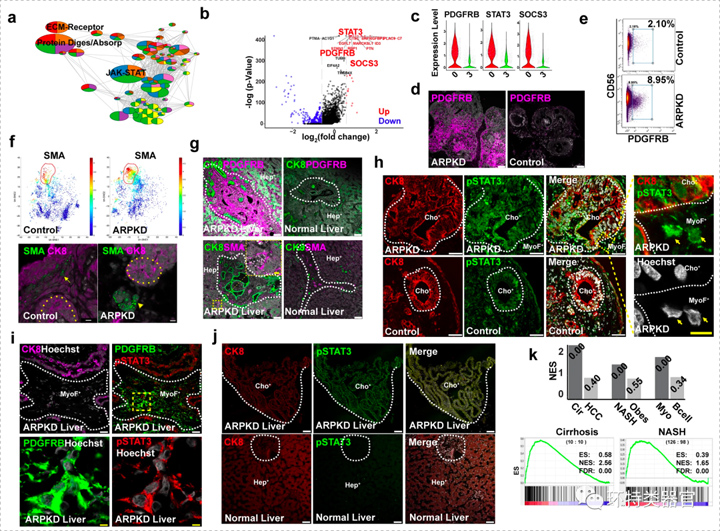
常染色体隐性多囊肾(ARPKD)是一种单基因疾病,可导致肾脏和肝脏病理异常,病发后期肝脏病变为其主要致死病因[10, 11]。ARPKD肝病特征为肝内胆管扩张和胆道纤维化,即先天性肝纤维化(CHF)[11]。在本研究中,研究人员对人源多能干细胞(iPSC)分化的肝脏类器官进行基因编辑,引起表达ARPKD最常见的致病突变。结果显示这些肝类器官仅在21天内就发展出ARPKD肝脏病变的关键特征,其肌成纤维细胞的转录组与常见肝纤维化患者肝组织的转录组相似。ARPKD类器官为先天性(和可能获得性)肝纤维化发病机制及药物筛选提供了强有力的研究模型。
一、主要研究结果
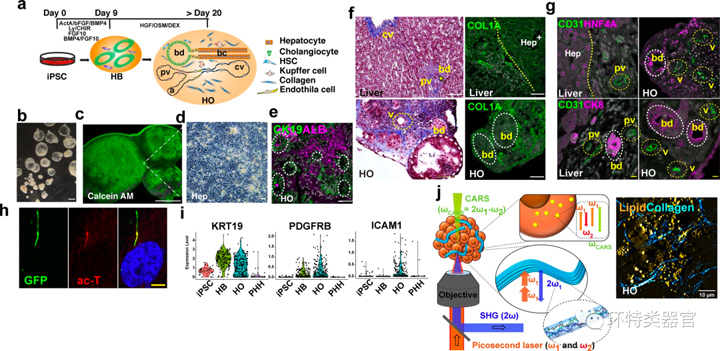
1. 肝脏类器官(HO)的表征
对HOs进行切片染色和单细胞测序,结果显示生成了肝细胞薄片和胆管细胞,且间充质组织与肝脏内的间充质组织相似,具有更复杂的抗原表达以及形成交联胶原纤维所需的酶学机制。
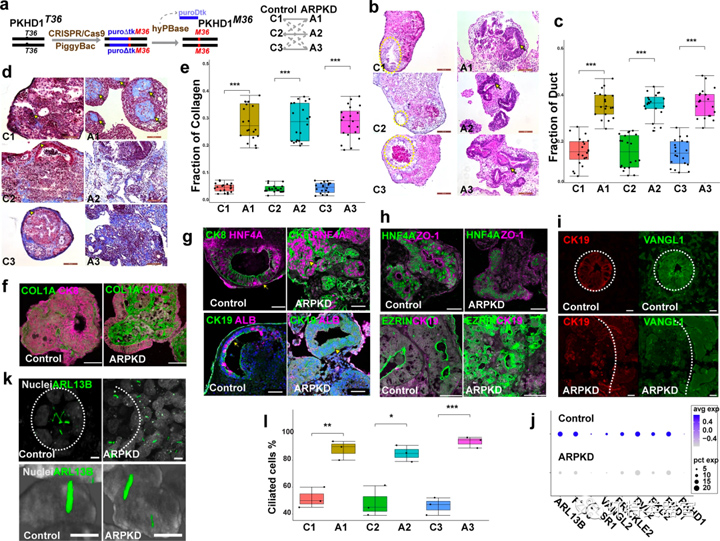
2. ARPKD肝脏病理学的类器官模型
对iPSC进行ARPKD突变并构建HOs,结果表明ARPKD类器官的不规则胆管数量较对照HOs增加,ECM数量也显著增加。免疫染色结果证实,COL1A数量增加,在ARPKD类器官中弥漫性沉积,且与初级纤毛或平面细胞极性相关的多种mRNA的表达水平降低,胆管细胞的极性被破坏。
在分化的不同阶段测量初级纤毛的数量和长度,发现ARPKD类器官中初级纤毛的相比对照组高约2倍,但长度没有明显变化,因此ARPKD可能不影响初级纤毛的长度,但会影响其功能。
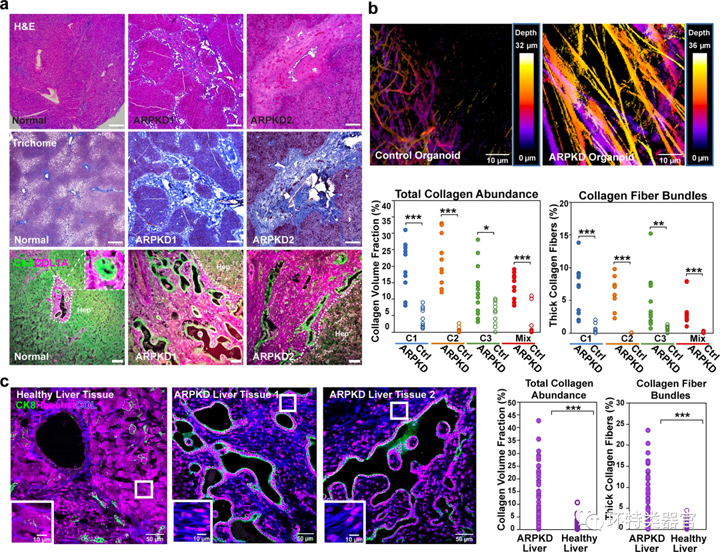
3. 纤维化定量
采用三维SHG定量评价对照组与ARPKD类器官中胶原蛋白纤维的丰度和结构,结果表明ARPKD类器官中肝组织胆管扩大、ECM含量显著增加,沉积在整个组织中,其胶原纤维丰度显著高于对照组。因此,ARPKD类器官中观察到的病理变化能够反映ARPKD肝脏组织中的变化。
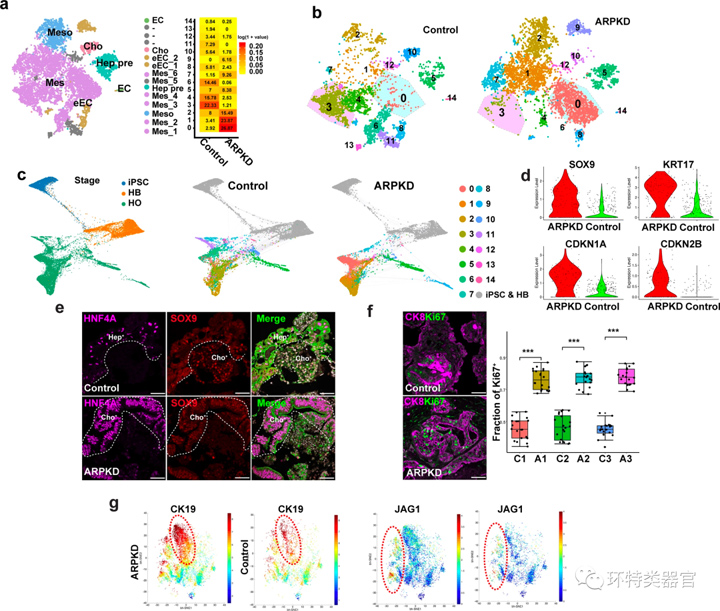
4. ARPKD的发病机制和对胆管细胞的影响
单细胞测序及转录组结果显示ARPKD类器官的细胞组成与对照组存在显著差异,ARPKD突变对间充质细胞的影响可能发生在肝细胞分化为成熟肝类器官时期。
此外,ARPKD类器官中的胆管细胞对调节细胞周期和有丝分裂的mRNA、胶原蛋白和参与胶原纤维生成的蛋白的基因的表达水平增加,其胆管细胞成熟度较低,但增殖能力较强,TGFβ通路激活增加。因此,相比对照组ARPKD类器官的胆管细胞会更加积极参与胶原纤维的生成。
5. ARPKD中肌成纤维细胞扩张
ARPKD突变也促进了具有活化和增殖细胞特征的肌成纤维细胞的产生,与纤维化的人类肝脏组织相似。
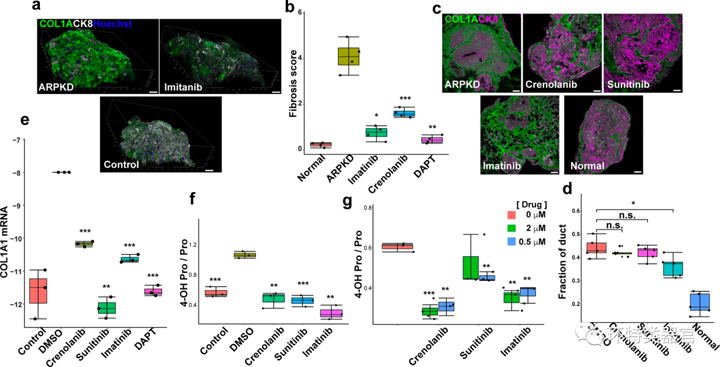
6. 抑制作用
为确定PDGFRB-STAT3通路是否与ARPKD纤维化的发病机制相关,采用三种方法对测定不同PDGFR酪氨酸激酶抑制剂(Crenolanib、Sunitinib和Imatinib)对ARPKD类器官纤维化的影响。
免疫染色结果表明,10 μM Imatinib和Crenolanib处理可显著降低ARPKD类器官中胶原蛋白的生成,对纤维化程度影响显著,表明PDGFRB-STAT3通路在肝纤维化的发病机制中起着重要作用。
原文献:Guan, Y., Enejder, A., Wang, M. et al. A human multi-lineage hepatic organoid model for liver fibrosis. Nat Commun 12, 6138 (2021).
参考资料:
[1] Yoon, Y. J., Friedman, S. L. & Lee, Y. A. Antifibrotic therapies: where are we now? Semin Liver Dis. 36, 87–98 (2016).
[2]. Hernandez-Gea, V. & Friedman, S. L. Pathogenesis of liver fibrosis. Annu. Rev.Pathol. 6, 425–456 (2011).
[3]. Zhang, C. Y., Yuan, W. G., He, P., Lei, J. H. & Wang, C. X. Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 22, 10512–10522 (2016).
[4]. Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation.Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
[5]. Yin, C., Evason, K. J., Asahina, K. & Stainier, D. Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 123, 1902–1910 (2013).
[6]. Zhang, D. Y. et al. A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut 65, 1754–1764 (2016).
[7]. Mederacke, I. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823 (2013).
[8]. Aycock, R. S. & Seyer, J. M. Collagens of normal and cirrhotic human liver. Connect Tissue Res. 23, 19–31 (1989).
[9]. Olaso, E. et al. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J. Clin. Invest. 108, 1369–1378 (2001).
[10]. Hartung, E. A. & Guay-Woodford, L. M. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics 134, e833–e845 (2014).
[11]. Buscher, R. et al. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr. Nephrol. 29, 1915–1925 (2014).
